 ,
, Статистическая физика, раздел физики, задача которого — выразить свойства макроскопических тел, т. е. систем, состоящих из очень большого числа одинаковых частиц (молекул, атомов, электронов и т.д.), через свойства этих частиц и взаимодействие между ними.
Изучением макроскопических тел занимаются и др. разделы физики — термодинамика, механика сплошных сред, электродинамика сплошных сред. Однако при решении конкретных задач методами этих дисциплин в соответствующие уравнения всегда входят неизвестные параметры или функции, характеризующие данное тело. Так, для решения задач гидродинамики необходимо знать уравнение состояния жидкости или газа, т. е. зависимость плотности от температуры и давления, теплоёмкость жидкости, её коэффициент вязкости и т.п. Все эти зависимости и параметры можно, разумеется, определять экспериментально, поэтому методы, о которых идёт речь, называются феноменологическими. Статистическая же физика позволяет, по крайней мере в принципе, а во многих случаях и фактически, вычислить все эти величины, если известны силы взаимодействия между молекулами. Т. о., С. ф. использует сведения о «микроскопическом» строении тел — о том, из каких частиц они состоят, как эти частицы взаимодействуют, поэтому её называют микроскопической теорией.
Если в какой-то момент времени заданы координаты и скорости всех частиц тела и известен закон их взаимодействия, то, решая уравнения механики, можно было бы найти эти координаты и скорости в любой последующий момент времени и тем самым полностью определить состояние исследуемого тела. (Для простоты изложение ведётся на языке классической механики. Но и в квантовой механике ситуация та же: зная начальную волновую функцию системы и закон взаимодействия частиц, можно, решая Шрёдингера уравнение, найти волновую функцию, определяющую состояние системы во все будущие моменты времени.) Фактически, однако, такой путь построения микроскопической теории невозможен, т.к. число частиц в макроскопических телах очень велико. Например, в 1 см3 газа при температуре 0 °С и давлении в 1 атм содержится примерно 2,7×1019 молекул. Невозможно решить такое число уравнений, а начальные координаты и скорости всех молекул всё равно неизвестны.
Однако именно большое число частиц в макроскопических телах приводит к появлению новых — статистических — закономерностей в поведении таких тел. Это поведение в широких пределах не зависит от конкретных начальных условий — от точных значений начальных координат и скоростей частиц. Важнейшее проявление этой независимости — известный из опыта факт, что система, предоставленная самой себе, т. е. изолированная от внешних воздействий, с течением времени приходит в некоторое равновесное состояние (термодинамическое, или статистическое, равновесие), свойства которого определяются только такими общими характеристиками начального состояния, как число частиц, их суммарная энергия и т.п. (см. Равновесие термодинамическое). В дальнейшем речь будет идти главным образом о С. ф. равновесных состояний.
Прежде чем сформулировать теорию, описывающую статистические закономерности, следует разумно ограничить сами требования к теории. Именно, задачей теории должно являться вычисление не точных значений различных физических величин для макроскопических тел, а средних значений этих величин по времени. Рассмотрим, например, молекулы, находящиеся в некотором выделенном в газе достаточно большом — макроскопическом — объёме. Число таких молекул с течением времени будет меняться из-за их движения, и его можно было бы найти точно, если были бы известны все координаты молекул во все моменты времени. В этом, однако, нет необходимости. Изменение числа молекул в объёме будет носить характер беспорядочных колебаний — флуктуаций — относительно некоторого среднего значения. При большом числе частиц в объёме эти колебания будут малы по сравнению со средним числом частиц, так что для характеристики макроскопического состояния достаточно знать именно это среднее значение.
Для уяснения характера статистических закономерностей рассмотрим ещё один простой пример. Пусть в некоторый сосуд помещено большое число зёрен двух сортов, каждого сорта поровну, и содержимое сосуда тщательно перемешано. Тогда на основании повседневного опыта можно быть уверенным, что во взятой из сосуда пробе, содержащей всё ещё большое число зёрен, будет обнаружено примерно равное число зёрен каждого сорта независимо от того, в каком порядке засыпались зёрна в сосуд. На этом примере хорошо видны два важных обстоятельства, обеспечивающих применимость статистической теории. Во первых, необходимость большого числа зёрен как во всей «системе» — сосуде с зерном, так и в выбранной для опыта «подсистеме» — пробе. (Если проба состоит всего из двух зёрен, то нередко оба будут одного сорта.) Во-вторых, ясно, что существенную роль играет сложность движения зёрен при перемешивании, обеспечивающая их равномерное распределение в объёме сосуда.
Функция распределения. Рассмотрим систему, состоящую из N частиц, для простоты считая, что частицы не имеют внутренних степеней свободы. Такая система описывается заданием 6N переменных — 3N координат qi и 3N импульсов pi, частиц [совокупность этих переменных сокращённо будет обозначаться (р, q)]. Вычислим среднее значение по интервалу временит некоторой величины F (р, q), являющейся функцией этих координат и импульсов. Для этого разобьем интервал (0, t) на s равных малых отрезков Dta (а = 1,2,....... s). Тогда по определению
,
или (1)
 ,
,
где qa и pa — значения координат и импульсов в моменты времени ta. В пределе s ® ¥ сумма переходит в интеграл:
 (1a)
(1a)
Понятие функции распределения естественным образом, возникает, если рассмотреть пространство 6N измерений, на осях которого отложены значения координат и импульсов частиц системы; оно называется фазовым пространством. Каждому значению времени t соответствуют определённые значения всех q и р, т. е. некоторая точка в фазовом пространстве, изображающая состояние системы в данный момент времени t. Разобьем всё фазовое пространство на элементы, размер которых мал по сравнению с характерными для данного состояния системы значениями q и р, но ещё настолько велик, что в каждом из них находится много точек, изображающих состояние системы в различные моменты времени t. Тогда число таких точек в элементе объёма будет примерно пропорционально величине этого объёма dpdq. Если обозначить коэффициент пропорциональности через sw(p, q), то это число для элемента с центром в некоторой точке (р, q) запишется в виде:
da = sw (р, q) dpdq, (2)
где
dpdq = dp1dq1dp2dq2... dp3Ndq3N
— объём выбранного элемента фазового пространства. Среднее значение (1) с учётом малости этих элементов объёма можно переписать как  , т. е.
, т. е.
 (3)
(3)
(интегрирование по координатам производится по всему объёму системы, по импульсам — от —¥ до ¥). Функция w(p, q, t) носит название функции распределения по координатами импульсам частиц. Поскольку полное число выбранных точек равно s, функция w удовлетворяет условию нормировки:
 (4)
(4)
Из (3) и (4) видно, что wdpdq можно рассматривать как вероятность системе находиться в элементе dpdq фазового пространства. Введённой таким образом функции распределения можно дать и др. истолкование. Для этого будем рассматривать одновременно большое число одинаковых систем и примем, что каждая точка в фазовом пространстве изображает состояние одной такой системы. Тогда усреднение по времени в (1)—(1a) можно понимать как усреднение по совокупности этих систем, или, как говорят, по статистическому ансамблю. Проведённые до сих пор рассуждения носили чисто формальный характер, т.к. нахождение функции распределения, согласно (2), требует знания всех р и q во все моменты времени, т. е. решения уравнений движения с соответствующими начальными условиями. Основным положением С. ф. является, однако, утверждение о возможности определить эту функцию из общих соображений для системы, находящейся в состоянии термодинамического равновесия. Прежде всего можно показать, исходя из сохранения числа систем при движении, что функция распределения является интегралом движения системы, т. е. остаётся постоянной, если р и q меняются в соответствии с уравнениями движения (см. Лиувилля теорема). При движении замкнутой системы не меняется её энергия, поэтому все точки в фазовом пространстве, изображающие состояние системы в разные моменты времени, должны лежать на некоторой «гиперповерхности», соответствующей начальному значению энергии Е. Уравнение этой поверхности имеет вид;
H (p, q) = E,
где Н (р, q) — энергия системы, выраженная через координаты и импульсы, т. е. её функция Гамильтона. Далее, движение системы из многих частиц носит крайне запутанный характер. Поэтому с течением времени точки, описывающие состояние, распределятся по поверхности постоянной энергии равномерно, подобно тому как равномерно распределяются зёрна при перемешивании в сосуде в упомянутом выше примере (см. также Эргодическая гипотеза). Такое равномерное распределение по изоэнергетической поверхности описывается функцией распределения вида:
w(p, q) = Ad[H (p, q) — E], (5)
где d[Н (р, q) — E] — дельта-функция, отличная от нуля только при Н = Е, т. е. на этой поверхности, А — постоянная, определяемая из условия нормировки (4). Функция распределения (5), называется микроканонической, позволяет вычислять средние значения всех физических величин по формуле (3), не решая уравнений движения.
При выводе выражения (5) предполагалось, что единственная сохраняющаяся при движении системы величина, от которой зависит w, — это энергия системы. Разумеется, сохраняются также импульс и момент импульса, но эти величины можно исключить, предположив, что рассматриваемое тело заключено в неподвижный ящик, которому частицы могут отдавать импульс и момент.
Фактически обычно рассматриваются не замкнутые системы, а макроскопические тела, являющиеся макроскопически малыми частями, или подсистемами, какой-либо замкнутой системы. Функция распределения для подсистемы будет отлична от (5), но не будет зависеть от конкретного характера остальной части системы — т. н. термостата. Поэтому функцию распределения подсистемы можно определить, считая, например, что термостат состоит просто из N частиц идеального газа, координаты и импульсы которых будем обозначать через Q и Р, в отличие от обозначений q и р для подсистемы, тогда микроканоническое распределение:

Здесь Н (р, q) — функция Гамильтона подсистемы, М — масса частицы газа, а суммирование производится по всем составляющим импульсов всех частиц термостата. Чтобы найти функцию распределения для подсистемы, нужно проинтегрировать это выражение по координатам и импульсам частиц термостата. Если затем учесть, что число частиц в термостате много больше, чем в подсистеме, и устремить N ®¥, считая, что отношение E/N постоянно и равно 3/2kT, то для функции распределения подсистемы получится выражение:
 (6)
(6)
Величина T в этой формуле имеет смысл температуры, k = 1,38×10-16 эрг/град — постоянная Больцмана. [Условие E/N ® 3/2kT для газа в термостате соответствует, как и должно быть, формуле (13) для идеального газа; см. ниже.] Нормировочный коэффициент eF/kT определяется из условия нормировки (4):
 (6a)
(6a)
Распределение (6) называется каноническим распределением Гиббса, или просто каноническим распределением (см. Гиббса распределение), а величина Z — статистическим интегралом. В отличие от микроканонического распределения, энергия системы в распределении Гиббса не задана. Состояния системы сосредоточены в тонком, но конечной толщины слое вокруг энергетической поверхности, соответствующей среднему значению энергии, что означает возможность обмена энергией с термостатом. В остальном в применении к определённому макроскопическому телу оба распределения приводят по существу к одним и тем же результатам. Разница лишь в том, что при использовании микроканонического распределения все средние значения оказываются выраженными через энергию тела, а при использовании канонического распределения — через температуру. Если тело состоит из двух невзаимодействующих частей 1 и 2 с функциями Гамильтона H1 и H2, то для всего тела Н = H1 + H2 и, согласно (6), функция распределения тела разбивается на произведение функций распределения для каждой из частей, так что эти части оказываются статистически независимыми. Это требование вместе с теоремой Лиувилля можно положить в основу вывода распределения Гиббса, не обращаясь к микроканоническому распределению. Формула (6) справедлива для систем, которые описываются классической механикой.
В квантовой механике энергетический спектр системы конечного объёма дискретен. Вероятность подсистеме находиться в состоянии с энергией En даётся формулой, аналогичной (6):
 , (7)
, (7)
причем условие нормировки  можно переписать в виде:
можно переписать в виде:
 . (8)
. (8)
Величина Z называется статистической суммой системы; сумма в выражении (8) берётся по всем состояниям системы.
Для системы, с достаточной точностью описывающейся классической механикой, в формуле (8) можно перейти от суммирования по состояниям к интегрированию по координатам и импульсам системы, При этом на каждое квантовое состояние приходится в фазовом пространстве «клетка» (или «ячейка») объемом  , где
, где  — Планка постоянная. Иными словами, суммирование по n сводится к интегрированию по
— Планка постоянная. Иными словами, суммирование по n сводится к интегрированию по  . Следует также учесть, что ввиду тождественности частиц в квантовой механике при их перестановке состояние системы не меняется. Поэтому, если интегрировать по всем р и q, необходимо поделить интеграл на число перестановок из N частиц, т. е. на N! Окончательно классический предел для статистической суммы имеет вид:
. Следует также учесть, что ввиду тождественности частиц в квантовой механике при их перестановке состояние системы не меняется. Поэтому, если интегрировать по всем р и q, необходимо поделить интеграл на число перестановок из N частиц, т. е. на N! Окончательно классический предел для статистической суммы имеет вид:
 (8а)
(8а)
Он отличается множителем от чисто классического условия нормировки (6а), что приводит к дополнительному слагаемому в F.
Приведенные формулы относятся к случаю, когда число частиц в подсистеме задано. Если выбрать в качестве подсистемы определенный элемент объёма всей системы, через поверхность которого частицы могут покидать подсистему и возвращаться в неё, то вероятность нахождения подсистемы в состоянии с энергией En и числом частиц Nn даётся формулой большого канонического распределения Гиббса:
 , (9)
, (9)
в которой дополнительный параметр m — химический потенциал, определяющий среднее число частиц в подсистеме, а величина W определяется из условия нормировки [см. формулу (11)].
Статистическое истолкование термодинамики. Важнейший результат С. ф. — установление статистического смысла термодинамических величин. Это даёт возможность вывести законы термодинамики из основных представлений С. ф. и вычислять термодинамические величины для конкретных систем. Прежде всего термодинамическая внутренняя энергия отождествляется со средней энергией системы. Первое начало термодинамики получает тогда очевидное истолкование как выражение закона сохранения энергии при движении составляющих тело частиц.
Далее, пусть функция Гамильтона системы зависит от некоторого параметра l (координаты стенки сосуда, в который заключена система, внешнего поля и т.п.). Тогда производная  будет обобщённой силой, соответствующей этому параметру, а величина
будет обобщённой силой, соответствующей этому параметру, а величина  после усреднения даёт механическую работу, совершаемую над системой при изменении этого параметра. Если продифференцировать выражение
после усреднения даёт механическую работу, совершаемую над системой при изменении этого параметра. Если продифференцировать выражение  для средней энергии
для средней энергии  системы с учетом формулы (6) и условия нормировки, считая переменными l и T и учитывая, что величина F тоже является функцией от этих переменных, то получится тождество:
системы с учетом формулы (6) и условия нормировки, считая переменными l и T и учитывая, что величина F тоже является функцией от этих переменных, то получится тождество:
 .
.
Согласно сказанному выше, член, содержащий dl, равен средней работе dA, совершаемой над телом. Тогда последний член есть получаемое телом тепло. Сравнивая это выражение с соотношением dE = dA + TdS, представляющим собой объединённую запись первого и второго начал термодинамики (см. Второе начало термодинамики) для обратимых процессов, находим, что T в (6) действительно равна абсолютной температуре тела, а производная  — взятой с обратным знаком энтропии S. Это означает, что F есть свободная энергия тела, откуда выясняется её статистический смысл.
— взятой с обратным знаком энтропии S. Это означает, что F есть свободная энергия тела, откуда выясняется её статистический смысл.
Особое значение имеет статистическое истолкование энтропии, которое следует из формулы (8). Формально суммирование g этой формуле производится по всем состояниям с энергией En, но фактически ввиду малости флуктуаций энергии в распределении Гиббса существенно лишь относительно небольшое их число с энергией вблизи средней энергии. Число этих существенных состояний  естественно определить поэтому, ограничив суммирование в (8) интервалом
естественно определить поэтому, ограничив суммирование в (8) интервалом 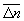 , заменив En на среднюю энергию
, заменив En на среднюю энергию  и вынося экспоненту из-под знака суммы. Тогда сумма даст
и вынося экспоненту из-под знака суммы. Тогда сумма даст  и примет вид.
и примет вид.

С др. стороны, согласно термодинамике, F =  — TS, что дает связь энтропии с числом микроскопических состояний
— TS, что дает связь энтропии с числом микроскопических состояний  в данном макроскопическом состоянии, иначе говоря, — со статистическим весом макроскопического состояния, т. е. с его вероятностью:
в данном макроскопическом состоянии, иначе говоря, — со статистическим весом макроскопического состояния, т. е. с его вероятностью:
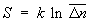 . (10)
. (10)
При температуре абсолютного нуля любая система находится в определённом основном состоянии, так что  = 1, S = 0. Это утверждение выражает собой третье начало термодинамики. Здесь существенно, что для однозначного определения энтропии нужно пользоваться именно квантовой формулой (8); в чисто классической статистике энтропия определена только с точностью до произвольного слагаемого.
= 1, S = 0. Это утверждение выражает собой третье начало термодинамики. Здесь существенно, что для однозначного определения энтропии нужно пользоваться именно квантовой формулой (8); в чисто классической статистике энтропия определена только с точностью до произвольного слагаемого.
Смысл энтропии как меры вероятности состояния сохраняется и по отношению к произвольным — не обязательно равновесным — состояниям. В состоянии равновесия энтропия имеет максимальное возможное в данных внешних условиях значение. Это означает, что равновесное состояние является состоянием с максимальным статистическим весом, наиболее вероятным состоянием. Процесс перехода системы из неравновесного состояния в равновесное есть процесс перехода из менее вероятных состояний в более вероятные; это выясняет статистический смысл закона возрастания энтропии, согласно которому энтропия замкнутой системы может только увеличиваться.
Формула (8), связывающая свободную энергию F со статистической суммой, является основой для вычисления термодинамических величин методами С. ф. Она используется, в частности, для построения статистической теории электрических и магнитных свойств вещества. Например, для вычисления магнитного момента тела в магнитном поле следует вычислить статистическую сумму и свободную энергию. Магнитный момент m тела дается тогда формулой:
m =  ,
,
где Н — напряженность внешнего магнитного поля. Аналогично (8) условие нормировки в большом каноническом распределении (9) определяет термодинамический потенциал W согласно формуле:
 . (11)
. (11)
Этот потенциал связан со свободной энергией соотношением:
 .
.
Приложения С. ф. к изучению тех или иных свойств конкретных систем сводятся по существу к приближённому вычислению статистической суммы с учётом специфических свойств системы.
Во многих случаях эта задача упрощается применением закона равнораспределения по степеням свободы, утверждающего, что теплоёмкость cv (при постоянном объёме v) системы взаимодействующих материальных точек — частиц, совершающих гармонические колебания, равна
cv = k (l/2 + n),
где l — общее число поступательных и вращательных степеней свободы, n — число колебательных степеней свободы. Доказательство закона основано на том, что функция Гамильтона Н такой системы имеет вид: Н = K (pi) + U (qm), где кинетическая энергия К — однородная квадратичная функция от l + n импульсов pi а потенциальная энергия U — квадратичная функция от n колебательных координат qm. В статистическом интеграле Z (8а) интегрирование по колебательным координатам ввиду быстрой сходимости интеграла можно распространить от - ¥ до ¥. Сделав после этого замену переменных  ,
,  находим, что Z зависит от температуры как T l/2+n, так что свободная энергия F = — kT (l/2 + n)(lnT+ const). Отсюда следует приведённое выше выражение для теплоёмкости, поскольку
находим, что Z зависит от температуры как T l/2+n, так что свободная энергия F = — kT (l/2 + n)(lnT+ const). Отсюда следует приведённое выше выражение для теплоёмкости, поскольку  . Отклонения от закона равнораспределения в реальных системах связаны прежде всего с квантовыми поправками, т.к. в квантовой С. ф. этот закон несправедлив. Существуют также поправки, связанные с негармоничностью колебаний.
. Отклонения от закона равнораспределения в реальных системах связаны прежде всего с квантовыми поправками, т.к. в квантовой С. ф. этот закон несправедлив. Существуют также поправки, связанные с негармоничностью колебаний.
Идеальный газ. Простейшим объектом исследования С. ф. является идеальный газ, т. е. газ настолько разреженный, что можно пренебречь взаимодействием между его молекулами. Термодинамические функции такого газа можно вычислить до конца. Энергия газа равна просто сумме энергий отдельных молекул. Этого, однако, ещё недостаточно, чтобы считать молекулы полностью независимыми. Действительно, в квантовой механике, даже если силы взаимодействия между частицами отсутствуют, существует определённое влияние одинаковых (тождественных) частиц друг на друга, если они находятся в близких квантовомеханических состояниях. Это т. н. обменное взаимодействие. Им можно пренебречь, если на одно состояние приходится в среднем много меньше одной частицы, что во всяком случае имеет место при достаточно высокой температуре газа; такой газ называется невырожденным. Фактически обычные газы, состоящие из атомов и молекул, невырождены при всех температурах (при которых они ещё газообразны). Для невырожденного идеального газа функция распределения распадается на произведение функций распределения для отдельных молекул. лежат в интервалах dpx, dpy, dpz, а координаты — в интервалах dx, dy, dz:
 , (12) Энергия молекулы одноатомного газа во внешнем поле с потенциальной энергией U (r) равна p2/2M + U (r). Интегрируя (6) по координатам r (x, у, z) и импульсам р (рх, py, pz) всех молекул, кроме одной, можно найти число молекул dN, импульсы которых
, (12) Энергия молекулы одноатомного газа во внешнем поле с потенциальной энергией U (r) равна p2/2M + U (r). Интегрируя (6) по координатам r (x, у, z) и импульсам р (рх, py, pz) всех молекул, кроме одной, можно найти число молекул dN, импульсы которых
где d3p = dpxdpydpz, d3x = dxdydz. Эта формула называется распределением Максвелла — Больцмана (см. Больцмана статистика). Если проинтегрировать (12) по импульсам, то получится формула для распределения частиц по координатам во внешнем поле, в частности в поле тяготения — барометрическая формула. Распределение же по скоростям в каждой точке пространства совпадает с Максвелла распределением.
Статистическая сумма идеального газа также распадается на произведение одинаковых членов, соответствующих отдельным молекулам. Для одноатомного газа суммирование в (8) сводится к интегрированию по координатам и импульсам, т. е. сумма заменяется на интеграл по  3 в соответствии с числом ячеек [с объёмом
3 в соответствии с числом ячеек [с объёмом  ] в фазовом пространстве одной частицы. Свободная энергия N атомов газа равна:
] в фазовом пространстве одной частицы. Свободная энергия N атомов газа равна:
 ,
,
где g — статистический вес основного состояния атома, т. е. число состояний, соответствующее его нижнему энергетическому уровню, V — объём газа (здесь е — основание натуральных логарифмов). При высоких температурах g = (2J + 1)(2L + 1), где J — величина спина, a L — момента орбитального атома (в единицах 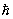 ). Из выражения для свободной энергии следует, что уравнение состояния идеального газа, т. е. зависимость его давления (Р) от плотности числа частиц (N/V) и температуры, имеет вид: PV = NkT. Внутренняя энергия одноатомного газа и его теплоёмкость при постоянном объёме оказываются равными:
). Из выражения для свободной энергии следует, что уравнение состояния идеального газа, т. е. зависимость его давления (Р) от плотности числа частиц (N/V) и температуры, имеет вид: PV = NkT. Внутренняя энергия одноатомного газа и его теплоёмкость при постоянном объёме оказываются равными:
Е = 3/2(NkT), Cv = 3/2Nk, (13)
а его химический потенциал:
 . (14)
. (14)
Характерно, что даже для невырожденного (т. е. с достаточной точностью подчиняющегося классической механике) газа выражения для свободной энергии и химического потенциала содержат постоянную Планка  . Это, в конечном счёте, обусловлено отмеченной ранее связью энтропии с понятием числа квантовых состояний.
. Это, в конечном счёте, обусловлено отмеченной ранее связью энтропии с понятием числа квантовых состояний.
В случае двухатомных и многоатомных газов вклад в термодинамические функции вносят также колебания и вращение молекул. Этот вклад зависит от того, существенны ли эффекты квантования колебаний и вращения молекулы. Расстояние между колебательными уровнями энергии имеет порядок  , где w — характерная частота колебаний, а расстояние между первыми вращательными уровнями энергии порядка
, где w — характерная частота колебаний, а расстояние между первыми вращательными уровнями энергии порядка  , где I — момент инерции вращающегося тела, в данном случае молекулы. Классическая статистика справедлива, если температура достаточно высока, так что
, где I — момент инерции вращающегося тела, в данном случае молекулы. Классическая статистика справедлива, если температура достаточно высока, так что
kT >> DE.
В этом случае в соответствии с законом равнораспределения вращение вносит в теплоёмкость постоянный вклад, равный 1/2k на каждую вращательную степень свободы; в частности, для двухатомных молекул этот вклад равен k. Колебания же вносят в теплоёмкость вклад, равный k на каждую колебательную степень свободы (так что колебательная теплоёмкость двухатомной молекулы равна k). Вдвое больший вклад колебательной степени свободы по сравнению с вращательной связан с тем, что при колебаниях атомы в молекуле имеют не только кинетическую, но и потенциальную энергию. В обратном предельном случае  молекулы находятся в своём основном колебательном состоянии, энергия которого не зависит от температуры, так что колебания вообще не вносят вклада в теплоёмкость. То же относится к вращению молекул при условии
молекулы находятся в своём основном колебательном состоянии, энергия которого не зависит от температуры, так что колебания вообще не вносят вклада в теплоёмкость. То же относится к вращению молекул при условии  . По мере повышения температуры появляются молекулы, находящиеся в возбуждённых колебательных и вращательных состояниях, и эти степени свободы начинают давать вклад в теплоёмкость — как бы постепенно «включаются», стремясь при дальнейшем повышении температуры к своему классическому пределу. Т. о., учёт квантовых эффектов позволил объяснить экспериментально наблюдаемую зависимость теплоёмкости газов от температуры. Значения величины
. По мере повышения температуры появляются молекулы, находящиеся в возбуждённых колебательных и вращательных состояниях, и эти степени свободы начинают давать вклад в теплоёмкость — как бы постепенно «включаются», стремясь при дальнейшем повышении температуры к своему классическому пределу. Т. о., учёт квантовых эффектов позволил объяснить экспериментально наблюдаемую зависимость теплоёмкости газов от температуры. Значения величины  , характеризующей «вращательный квант», для большинства молекул порядка нескольких градусов или десятков градусов (85 К для H2, 2,4 К для O2, 15 К для HCl). В то же время характерные значения величины
, характеризующей «вращательный квант», для большинства молекул порядка нескольких градусов или десятков градусов (85 К для H2, 2,4 К для O2, 15 К для HCl). В то же время характерные значения величины 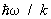 для «колебательного кванта» порядка тысяч градусов (6100 К для H2, 2700 К для O2, 4100 К для HCl). Поэтому вращательные степени свободы включаются при гораздо более низких температурах, чем колебательные. На рис. 1 изображены температурная зависимость вращательной (а) и колебательной (б) теплоёмкостей для двухатомной молекулы (вращательная теплоёмкость построена для молекулы из разных атомов).
для «колебательного кванта» порядка тысяч градусов (6100 К для H2, 2700 К для O2, 4100 К для HCl). Поэтому вращательные степени свободы включаются при гораздо более низких температурах, чем колебательные. На рис. 1 изображены температурная зависимость вращательной (а) и колебательной (б) теплоёмкостей для двухатомной молекулы (вращательная теплоёмкость построена для молекулы из разных атомов).
Неидеальный газ. Важное достижение С. ф. — вычисление поправок к термодинамическим величинам газа, связанных с взаимодействием между его частицами. С этой точки зрения уравнение состояния идеального газа является первым членом разложения давления реального газа по степеням плотности числа частиц, поскольку всякий газ при достаточно малой плотности ведёт себя как идеальный. С повышением плотности начинают играть роль поправки к уравнению состояния, связанные с взаимодействием. Они приводят к появлению в выражении для давления членов с более высокими степенями плотности числа частиц, так что давление изображается т. н. вириальным рядом вида:
 . (15)
. (15)
Коэффициенты В, С и т.д. зависят от температуры и наываются. вторым, третьим и т.д. вириальными коэффициентами. Методы С. ф. позволяют вычислить эти коэффициенты, если известен закон взаимодействия между молекулами газа. При этом коэффициенты В, С,... описывают одновременное взаимодействие двух, трёх и большего числа молекул. Например, если газ одноатомный и потенциальная энергия взаимодействия его атомов U (r), то второй вириальный коэффициент равен
 . (16)
. (16)
По порядку величины В равен  , где r0 — характерный размер атома, или, точнее, радиус действия межатомных сил. Это означает, что ряд (15) фактически представляет собой разложение по степеням безразмерного параметра Nr3/V, малого для достаточно разреженного газа. Взаимодействие между атомами газа носит характер отталкивания на близких расстояниях и притяжения на далёких. Это приводит к тому, что В > 0 при высоких температурах и В < 0 при низких. Поэтому давление реального газа при высоких температурах больше давления идеального газа той же плотности, а при низких — меньше. Так, например, для гелия при Т = 15,3 К коэффициент В = —3×10-23 см3, а при T = 510 К В = 1,8 ×10-23 см3. Для аргона В = —7,1×10-23 см3 при Т = 180 К и В = 4,2×10-23 см3 при Т = 6000 К. Для одноатомных газов вычислены значения вириальных коэффициентов, включая пятый, что позволяет описывать поведение газов в достаточно широком интервале плотностей (см. также Газы).
, где r0 — характерный размер атома, или, точнее, радиус действия межатомных сил. Это означает, что ряд (15) фактически представляет собой разложение по степеням безразмерного параметра Nr3/V, малого для достаточно разреженного газа. Взаимодействие между атомами газа носит характер отталкивания на близких расстояниях и притяжения на далёких. Это приводит к тому, что В > 0 при высоких температурах и В < 0 при низких. Поэтому давление реального газа при высоких температурах больше давления идеального газа той же плотности, а при низких — меньше. Так, например, для гелия при Т = 15,3 К коэффициент В = —3×10-23 см3, а при T = 510 К В = 1,8 ×10-23 см3. Для аргона В = —7,1×10-23 см3 при Т = 180 К и В = 4,2×10-23 см3 при Т = 6000 К. Для одноатомных газов вычислены значения вириальных коэффициентов, включая пятый, что позволяет описывать поведение газов в достаточно широком интервале плотностей (см. также Газы).
Плазма. Особый случай неидеального газа представляет собой плазма — частично или полностью ионизованный газ, в котором поэтому имеются свободные электроны и ионы. При достаточно малой плотности свойства плазмы близки к свойствам идеального газа. При вычислении же отклонений от идеальности существенно, что электроны и ионы взаимодействуют электростатически по закону Кулона. Кулоновские силы медленно убывают с расстоянием, и это приводит к тому, что уже для вычисления первой поправки к термодинамическим функциям необходимо учитывать взаимодействие не двух, а сразу большого количества частиц, поскольку интеграл во втором вириальном коэффициенте (16), описывающий парное взаимодействие, расходится на больших расстояниях r между частицами. В действительности под влиянием кулоновских сил распределение ионов и электронов в плазме изменяется таким образом, что поле каждой частицы экранируется, т. е. быстро убывает на некотором расстоянии, называемом дебаевским радиусом. Для простейшего случая плазмы, состоящей из электронов и однозарядных ионов, дебаевский радиус rD равен:
 , (17)
, (17)
где N число электронов, е — заряд электрона. Все частицы, находящиеся внутри дебаевского радиуса, принимают участие во взаимодействии одновременно. Это приводит к тому, что первая поправка к давлению пропорциональна не (N/V)2 как в обычном газе, а более низкой степени плотности — (N/V)3/2. Количественный расчёт основан на том, что остальные частицы распределены в поле выбранного электрона или иона согласно распределению Больцмана. В результате уравнение состояния с учётом первой поправки имеет вид:
 (18)
(18)
(т.к. число электронов равно числу ионов, полное число частиц равно 2N). Такого же рода поправки возникают и в термодинамических функциях электролитов, в которых имеются свободные ионы растворённых веществ.
Жидкости. В отличие от газа, связанные с взаимодействием члены в уравнении состояния жидкости не малы. Поэтому свойства жидкости сильно зависят от конкретного характера взаимодействия между её молекулами. В теории жидкости вообще отсутствует малый параметр, который можно было бы использовать для упрощения теории. Невозможно получить какие-либо аналитические формулы для термодинамических величин жидкости. Одним из способов преодоления этой трудности является изучение системы, состоящей из сравнительно небольшого числа частиц — порядка нескольких тысяч. В этом случае, используя ЭВМ, можно провести прямое решение уравнений движения частиц и определить таким способом средние значения всех характеризующих систему величин без дополнительных предположений. При этом можно исследовать также и процесс приближения такой системы к состоянию равновесия. Можно также найти статистический интеграл для такой системы из небольшого числа частиц путём вычисления на ЭВМ интегралов в основной формуле для статистического интеграла (обычно при этом используется Монте-Карло метод). Полученные обоими способами результаты имеют, однако, малую точность в приложении к реальным жидкостям из-за малого числа частиц в системе.
Ещё один способ построения теории жидкости основан на использовании функций распределения молекул. Если проинтегрировать функцию распределения w системы по импульсам всех частиц и по координатам всех частиц, кроме одной, получится одночастичная пространственная функция распределения f1(r). Если проинтегрировать w по импульсам всех частиц и по координатам всех частиц, кроме двух, получится двухчастичная функция распределения f2(r1, r2), всех частиц, кроме трёх, — трёхчастичная функция распределения f3(r1, r2, r3,) и т.д. Двухчастичная функция распределения является непосредственно наблюдаемой физической величиной — через неё выражается, например, упругое рассеяние рентгеновских лучей и нейтронов в жидкости. Считая, что функция распределения всей системы даётся распределением Гиббса (6), можно получить интегральное соотношение, выражающее двухчастичную функцию через трёхчастичную и потенциал взаимодействия между частицами. В теории жидкости это точное соотношение дополняется некоторыми приближёнными, выражающими трёхчастичную функцию через двухчастичную (одночастичная функция в однородной жидкости сводится к постоянной). В результате получается уравнение для двухчастичной функции, которое решается численно. Дополнительные соотношения находятся на основании правдоподобных физических соображений и носят интерполяционный характер, так что основанные на них теории могут претендовать лишь на качественное описание свойств жидкости. Тем не менее даже такое качественное описание имеет важное значение, поскольку в нём проявляется общность законов С. ф. (см. также Жидкость).
Химическое равновесие. Большое значение имеет предоставляемая С. ф. возможность вычисления констант химического равновесия, определяющих равновесные концентрации реагирующих веществ. Термодинамическая теория приводит к условию равновесия в виде равенства нулю некоторой линейной комбинации химических потенциалов этих веществ. В случае реакции между газами химические потенциалы определяются формулами, аналогичными формуле (14) для одноатомного газа, и константу равновесия можно вычислить, если известна теплота реакции. В выражения для химических потенциалов входит постоянная Планка, поэтому квантовые эффекты существенны даже для реакций между классическими газами. Важным частным случаем формул химического равновесия является Саха формула, определяющая равновесную степень ионизации газа. (Подробнее см. Равновесие химическое.)
Вырожденные газы. Если понижать температуру газа при постоянной плотности, начинают проявляться квантово-механические эффекты, связанные со свойствами симметрии волновых функций системы одинаковых частиц. Газ «вырождается» (см. Вырожденный газ). Для частиц с полуцелым спином волновая функция должна менять знак при перестановке любой пары частиц. Это, в частности, приводит к тому, что в одном квантовом состоянии не может находиться больше одной частицы (Паули принцип). Количество частиц с целым спином в одном состоянии может быть любым, но требуемая в этом случае неизменность волновой функции при перестановке частиц и здесь приводит к изменению статистических свойств газа. Частицы с полуцелым спином описываются статистикой Ферми — Дирака; их называют фермионами. К фермионам относятся, например, электроны, протоны, нейтроны, атомы дейтерия, атомы лёгкого изотопа гелия 3Не. Частицы с целым спином — бозоны — описываются статистикой Бозе — Эйнштейна. К ним относятся атомы водорода, атомы 4Не, кванты света — фотоны.
Пусть среднее число частиц газа в единице объёма с импульсами, лежащими в интервале d3p, есть  , так что np — число частиц в одной ячейке фазового пространства (g = 2J + 1, где J — спин частицы). Тогда из распределения Гиббса следует, что для идеальных газов фермионов (верхний знак) и бозонов (нижний знак):
, так что np — число частиц в одной ячейке фазового пространства (g = 2J + 1, где J — спин частицы). Тогда из распределения Гиббса следует, что для идеальных газов фермионов (верхний знак) и бозонов (нижний знак):
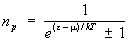 . (19)
. (19)
В этой формуле e = p2/2M — энергия частицы с импульсом р, m — химический потенциал, определяемый из условия постоянства числа частиц (N) в системе:
 .
.
Формула (19) переходит в формулу распределения Больцмана (12) при  ; левая сторона этого неравенства делается порядка правой при таких температурах, при которых длина волны де Бройля частиц, движущихся с тепловой скоростью, становится порядка среднего расстояния между ними. Т. о., вырождение сказывается при температурах тем более низких, чем меньше плотность числа частиц в газе (и чем больше масса частицы М).
; левая сторона этого неравенства делается порядка правой при таких температурах, при которых длина волны де Бройля частиц, движущихся с тепловой скоростью, становится порядка среднего расстояния между ними. Т. о., вырождение сказывается при температурах тем более низких, чем меньше плотность числа частиц в газе (и чем больше масса частицы М).
В случае фермионов, как и должно быть, np £ 1. Это приводит к тому, что частицы газа фермионов (ферми-газа) и при Т = 0 обладают отличными от нуля импульсами, поскольку в состоянии с нулевым импульсом может находиться только одна частица. Точнее, при Т = 0 для ферми-газа np = 1 внутри Ферми поверхности — сферы в импульсном пространстве с радиусом  , а вне этой «ферми-сферы» np = 0. При конечных, но низких температурах np меняется от 1 внутри сферы до нуля вне сферы постепенно, причём ширина переходной области порядка MkT/pF. Величина np для ферми-газа как функция от энергии e изображена схематически на рис. 2 (e0 = pF2/2M). При изменении температуры газа меняется состояние частиц только в этом переходном слое, и теплоёмкость ферми-газа при низких температурах пропорциональна Т и равна:
, а вне этой «ферми-сферы» np = 0. При конечных, но низких температурах np меняется от 1 внутри сферы до нуля вне сферы постепенно, причём ширина переходной области порядка MkT/pF. Величина np для ферми-газа как функция от энергии e изображена схематически на рис. 2 (e0 = pF2/2M). При изменении температуры газа меняется состояние частиц только в этом переходном слое, и теплоёмкость ферми-газа при низких температурах пропорциональна Т и равна:
 . (20)
. (20)
В бозе-газе при Т = 0 все частицы находятся в состоянии с нулевым импульсом. При достаточно низких температурах в состоянии с р = 0 находится конечная доля всех частиц; эти частицы образуют т. н. бозе-эйнштейновский конденсат. Остальные частицы находятся в состояниях с р ¹ 0, причём их число определяется формулой (19) с m = 0. При температуре  в бозе-газе происходит фазовый переход (см. ниже). Доля частиц с нулевым импульсом обращается в нуль Бозе — Эйнштейна конденсация исчезает. Кривая зависимости теплоёмкости от температуры имеет в точке Tc излом. Распределение частиц по импульсам при Т > Тс даётся формулой (19) причём m < 0. Схематически функции распределения Максвелла, Ферми — Дирака и Бозе — Эйнштейна (при Т > Тс) изображены на рис 3.
в бозе-газе происходит фазовый переход (см. ниже). Доля частиц с нулевым импульсом обращается в нуль Бозе — Эйнштейна конденсация исчезает. Кривая зависимости теплоёмкости от температуры имеет в точке Tc излом. Распределение частиц по импульсам при Т > Тс даётся формулой (19) причём m < 0. Схематически функции распределения Максвелла, Ферми — Дирака и Бозе — Эйнштейна (при Т > Тс) изображены на рис 3.
Особым случаем применения статистики Бозе — Эйнштейна является равновесное электромагнитное излучение, которое можно рассматривать как газ, состоящий из фотонов. Энергия фотона связана с его импульсом соотношением  , где с — скорость света в вакууме. Число фотонов не является заданной величиной, а само определяется из условия термодинамического равновесия, поэтому их распределение по импульсам даётся формулой (19) с m = 0 (причём e= рс). Распределение энергии в спектре излучения получается умножением числа фотонов на энергию e, так что плотность энергии в интервале частот dw равна
, где с — скорость света в вакууме. Число фотонов не является заданной величиной, а само определяется из условия термодинамического равновесия, поэтому их распределение по импульсам даётся формулой (19) с m = 0 (причём e= рс). Распределение энергии в спектре излучения получается умножением числа фотонов на энергию e, так что плотность энергии в интервале частот dw равна  , причем np берётся при
, причем np берётся при  . Т. о. получается формула Планка для спектра равновесного (чёрного) излучения (см. Планка закон излучения).
. Т. о. получается формула Планка для спектра равновесного (чёрного) излучения (см. Планка закон излучения).
Кристаллическая решётка. Применение С. ф. к вычислению термодинамических функций кристаллической решётки основано на том, что атомы в решётке совершают малые колебания около своих положений равновесия. Это позволяет рассматривать решётку как совокупность связанных гармонических осцилляторов. В такой системе могут распространяться волны, характеризующиеся своим законом дисперсии, т. е. зависимостью частоты w от волнового вектора k. В квантовой механике эти волны можно рассматривать как совокупность т. н. элементарных возбуждений, или квазичастиц — фононов, обладающих энергией  и квазиимпульсом ћk. Основное отличие квазиимпульса от импульса состоит в том, что энергия фонона является периодической функцией квазиимпульса с периодом, по порядку величины равным
и квазиимпульсом ћk. Основное отличие квазиимпульса от импульса состоит в том, что энергия фонона является периодической функцией квазиимпульса с периодом, по порядку величины равным  , где а — постоянная решётки. Функция распределения фононов по квазиимпульсам даётся формулой распределения Бозе—Эйнштейна (19) с m = 0. При этом
, где а — постоянная решётки. Функция распределения фононов по квазиимпульсам даётся формулой распределения Бозе—Эйнштейна (19) с m = 0. При этом  . Т. о., знание зависимости w(k) позволяет вычислить теплоёмкость решётки. Эту зависимость можно определить из опытов по неупругому рассеянию нейтронов в кристалле (см. Нейтронография) или вычислить теоретически, задавая значения «силовых констант», определяющих взаимодействие атомов в решётке. При низких температурах существенны только фононы с малой частотой, соответствующие квантам обычных звуковых волн, для которых связь w с k линейна. Это приводит к тому, что теплоёмкость кристаллической решётки пропорциональна T3. При высоких температурах можно пользоваться законом равного распределения энергии по степеням свободы, так что теплоёмкость не зависит от температуры и равна 3Nk, где N — число атомов в кристалле.
. Т. о., знание зависимости w(k) позволяет вычислить теплоёмкость решётки. Эту зависимость можно определить из опытов по неупругому рассеянию нейтронов в кристалле (см. Нейтронография) или вычислить теоретически, задавая значения «силовых констант», определяющих взаимодействие атомов в решётке. При низких температурах существенны только фононы с малой частотой, соответствующие квантам обычных звуковых волн, для которых связь w с k линейна. Это приводит к тому, что теплоёмкость кристаллической решётки пропорциональна T3. При высоких температурах можно пользоваться законом равного распределения энергии по степеням свободы, так что теплоёмкость не зависит от температуры и равна 3Nk, где N — число атомов в кристалле.
Металлы. В металлах вклад в термодинамические функции дают также электроны проводимости. Состояние электрона в металле характеризуется квазиимпульсом, и, т.к. электроны подчиняются статистике Ферми — Дирака, их распределение по квазиимпульсам даётся формулой (19). Поэтому теплоёмкость электронного газа, а следовательно, и всего металла при достаточно низких температурах пропорциональна Т. Отличие от ферми-газа свободных частиц состоит в том, что поверхность Ферми, около которой сосредоточены «активные» электроны, уже не является сферой, а представляет собой некоторую сложную поверхность в пространстве квазиимпульсов. Форму поверхности Ферми, равно как и зависимость энергии от квазиимпульса вблизи этой поверхности, можно определять экспериментально, главным образом исследуя магнитные свойства металлов, а также рассчитывать теоретически, используя т. н. модель квазипотенциала. В сверхпроводниках (см. Сверхпроводимость) возбужденные состояния электрона отделены от ферми-поверхности щелью конечной ширины, что приводит к экспоненциальной зависимости электронной теплоёмкости от температуры. В ферромагнитных и антиферромагнитных веществах вклад в термодинамические функции дают также колебания магнитных моментов — спиновые волны.
В диэлектриках и полупроводниках при Т = 0 свободные электроны отсутствуют. При конечных температурах в них появляются заряженные квазичастицы — электроны с отрицательным зарядом и (в равном числе) «дырки» с положительным зарядом, Электрон и дырка могут образовать связанное состояние — квазичастицу, называемую экситоном. Др. тип экситона представляет собой возбуждённое состояние атома диэлектрика, перемещающееся в кристаллической решётке.
Методы квантовой теории поля в С. ф. При решении задач квантовой С. ф., прежде всего при исследовании свойств квантовых жидкостей, электронов в металлах и магнетиков, важное значение имеют методы квантовой теории поля, введённые в С. ф. сравнительно недавно. Основную роль в этих методах играет функция Грина G макроскопической системы, аналогичная функции Грина в квантовой теории поля. Она зависит от энергии e и импульса р, закон дисперсии квазичастиц e(р) определяется из уравнения:
 , (21)
, (21)
т. е. энергия квазичастицы определяется полюсом функции Грина. Существует регулярный метод вычисления функций Грина в виде ряда по степеням энергии взаимодействия между частицами. Каждый член этого ряда содержит многократные интегралы по энергиям и импульсам от функций Грина невзаимодействующих частиц и может быть изображен графически в виде диаграмм, аналогичных Фейнмана диаграммам в квантовой электродинамике. Каждая из этих диаграмм имеет определённый физический смысл, что позволяет отделить в бесконечном ряду члены, ответственные за интересующее явление, и просуммировать их. Существует также диаграммная техника для вычисления температурных функций Грина, позволяющих вычислять термодинамические величины непосредственно, без введения квазичастиц. Упомянутые в разделе о жидкости методы, использующие многочастичные функции распределения квазичастиц, во многих отношениях близки к методам квантовой теории поля. Использование этих функций всегда основано на приближённом «расцеплении» — выражении функции более высокого порядка через функции более низкого.
Фазовые переходы. При непрерывном изменении внешних параметров (например, давления или температуры) свойства системы могут при некоторых значениях параметров измениться скачкообразно, т. е. происходит фазовый переход. Фазовые переходы делятся на переходы первого рода, сопровождающиеся выделением скрытой теплоты перехода и скачкообразным изменением объёма (к ним относится, например, плавление), и переходы второго рода, в которых скрытая теплота и скачок объёма отсутствуют (например, переход в сверхпроводящее состояние). Статистическая теория фазовых переходов составляет важную, но ещё далёкую от завершения область С. ф. Наибольшую трудность для теоретического исследования представляют при этом свойства вещества вблизи линии фазового перехода второго рода и вблизи критической точки фазового перехода первого рода. С математической точки зрения термодинамические функции системы имеют здесь особенности. Вблизи этих точек происходят своеобразные критические явления. В то же время здесь аномально возрастают флуктуации, и рассмотренные выше приближённые методы С. ф. оказываются неприменимыми. Поэтому важную роль играет небольшое число точно решаемых моделей, в которых есть переходы (например, т. н. модель Изинга).
Флуктуации. В основе С. ф. лежит тот факт, что физические величины, характеризующие макроскопические тела, с большой точностью равны своим средним значениям. Это равенство является всё же приближённым, в действительности все величины испытывают малые беспорядочные отклонения от средних значений — флуктуации. Существование флуктуаций имеет большое принципиальное значение, т.к. прямо доказывает статистический характер термодинамических закономерностей. Кроме того, флуктуации играют роль шума, мешающего физическим измерениям и ограничивающего их точность. Флуктуации некоторой величины х около её среднего значения  характеризуются средним квадратом флуктуации
характеризуются средним квадратом флуктуации
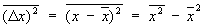 .
.
В подавляющем большинстве случаев величина х испытывает флуктуации порядка , существенно большие флуктуации встречаются крайне редко. Знание функции распределения системы позволяет вычислить средний квадрат флуктуации точно так же, как и среднее значение любой физической величины. Малые флуктуации термодинамических величин можно вычислить, используя статистическое истолкование энтропии. Согласно (10), вероятность неравновесного состояния системы с энтропией S пропорциональна eS/k. Это приводит к формуле
, существенно большие флуктуации встречаются крайне редко. Знание функции распределения системы позволяет вычислить средний квадрат флуктуации точно так же, как и среднее значение любой физической величины. Малые флуктуации термодинамических величин можно вычислить, используя статистическое истолкование энтропии. Согласно (10), вероятность неравновесного состояния системы с энтропией S пропорциональна eS/k. Это приводит к формуле
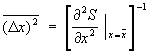 . (22)
. (22)
Например, средние квадраты флуктуаций объёма и температуры тела равны:
 ,
,  (23)
(23)
Из этих формул видно, что относительные флуктуации объёма и флуктуации температуры обратно пропорциональны  , где N — число частиц в теле. Это и обеспечивает малость флуктуаций для макроскопических тел. Связь между флуктуациями различных величин xi, xk характеризуется функцией
, где N — число частиц в теле. Это и обеспечивает малость флуктуаций для макроскопических тел. Связь между флуктуациями различных величин xi, xk характеризуется функцией  . Если флуктуации величин xi и xk статистически независимы, то
. Если флуктуации величин xi и xk статистически независимы, то  .
.
Под xi и xk можно понимать и значения одной и той же величины, например плотности, в различных точках пространства. Тогда эта функция имеет смысл пространственной корреляционной функции. С увеличением расстояния между точками корреляционная функция стремится к нулю (обычно экспоненциально), т.к. флуктуации в далёких точках пространства происходят независимо. Расстояние, на котором эта функция существенно убывает, называется корреляционным радиусом.
Временной ход флуктуаций и спектральное распределение флуктуационного шума описываются временной корреляционной функцией j(t), в которой усредняются флуктуации величины, взятые в различные моменты времени t:

Важную роль в теории флуктуаций играет т. н. флуктуационно-диссипативная теорема, связывающая флуктуации в системе с изменением её свойств под влиянием определённых внешних воздействий. Простейшее соотношение такого рода можно получить, рассматривая флуктуации гармонического осциллятора с потенциальной энергией  , где m — масса осциллятора, w0 — его собственная частота. Вычисление с помощью формулы (22) даёт:
, где m — масса осциллятора, w0 — его собственная частота. Вычисление с помощью формулы (22) даёт: 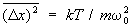 . С др. стороны, если на осциллятор действует сила f, среднее значение
. С др. стороны, если на осциллятор действует сила f, среднее значение  смещается на величину
смещается на величину  , так что
, так что
 (24)
(24)
и флуктуация х действительно связана с возмущением под влиянием силы f. В общем случае флуктуационно-диссипативная теорема применима, если для х существует «обобщённая сила» f, которая входит в оператор энергии системы (гамильтониан; см. Квантовая механика) в виде члена  , где
, где  — квантовомеханический оператор, соответствующий величине х. Включение силы f приведёт к изменению среднего значения
— квантовомеханический оператор, соответствующий величине х. Включение силы f приведёт к изменению среднего значения  на величину d
на величину d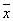 , причём, если f зависит от времени как е-iwt, это изменение можно записать в виде:
, причём, если f зависит от времени как е-iwt, это изменение можно записать в виде:
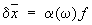 ;
;
комплексная величина a(w) называется обобщённой восприимчивостью системы. Теорема утверждает, что фурье-образ корреляционной функции

выражается через a следующим образом:
 (25)
(25)
(Im означает мнимую часть функции). Частным случаем (25) является Найквиста формула.
С. ф. неравновесных процессов. Всё большее значение приобретает кинетика физическая — раздел С. ф., изучающий процессы в системах, находящихся в неравновесных состояниях. Здесь возможны две постановки вопроса. Во-первых, можно рассматривать систему в некотором неравновесном состоянии и следить за её переходом в состояние равновесия. Во-вторых, можно рассматривать систему, неравновесное состояние которой поддерживается внешними условиями, например тело, в котором задан градиент температуры, протекает электрический ток и т.п., или тело, находящееся в переменном внешнем поле.
Если отклонение от равновесия мало, неравновесные свойства системы описываются т. н. кинетическими коэффициентами. Примерами таких коэффициентов являются коэффициенты вязкости, теплопроводности и диффузии, электропроводность металлов и т.п. Эти величины удовлетворяют принципу симметрии кинетических коэффициентов, выражающему симметрию уравнений механики относительно изменения знака времени (см. Онсагера теорема). В силу этого принципа, например, электропроводность кристалла описывается симметричным тензором.
Описание сильно неравновесных состояний, а также вычисление кинетических коэффициентов производятся с помощью кинетического уравнения. Это уравнение представляет собой интегро-дифференциальное уравнение для одночастичной функции распределения (в квантовом случае — для одночастичной матрицы плотности, или статистического оператора). Такое замкнутое, т. е. не содержащее др. величин, уравнение невозможно получить в общем виде. При его выводе необходимо использовать малые параметры, имеющиеся в данной конкретной задаче. Важнейшим примером является кинетическое уравнение Больцмана, описывающее установление равновесия в газе за счёт столкновений между молекулами. Оно справедливо для достаточно разреженных газов, когда длина свободного пробега велика по сравнению с расстояниями между молекулами. Конкретный вид этого уравнения зависит от эффективного сечения рассеяния молекул друг на друге. Если это сечение известно, уравнение можно решать, разлагая искомую функцию по ортогональным полиномам (см. Ортогональная система функций). Таким способом можно вычислить кинетические коэффициенты газа, исходя из известных законов взаимодействия между молекулами. Уравнение Больцмана учитывает только парные столкновения между молекулами и описывает только первый неисчезающий член разложения этих коэффициентов по плотности газа. Удалось найти и более точное уравнение, учитывающее также тройные столкновения, что позволило вычислить следующий член разложения.
Особую проблему представляет вывод кинетического уравнения для плазмы. Из-за медленного убывания кулоновских сил с расстоянием даже при рассмотрении парных столкновений существенно экранирование этих сил остальными частицами.
Неравновесные состояния твёрдых тел и квантовых жидкостей можно при низких температурах рассматривать как неравновесные состояния газа соответствующих квазичастиц. Поэтому кинетические процессы в таких системах описываются кинетическими уравнениями для квазичастиц, учитывающими столкновения между ними и процессы их взаимного превращения.
Новые возможности открыло применение в физической кинетике методов квантовой теории поля. Кинетические коэффициенты системы можно выразить через её функцию Грина, для которой существует общий способ вычисления с помощью диаграмм. Это позволяет в ряде случаев получить кинетические коэффициенты без явного использования кинетического уравнения и исследовать неравновесные свойства системы, даже когда не выполняются условия применимости кинетического уравнения.
Основные вехи развития С. ф. С. ф. целиком основана на представлениях об атомном строении материи. Поэтому начальный период развития С. ф. совпадает с развитием атомистических представлений (см. Атомизм). Развитие С. ф. как раздела теоретической физики началось в середине 19 в. В 1859 Дж. Максвелл определил функцию распределения молекул газа по скоростям. В 1860—70 Р. Клаузиус ввёл понятие длины свободного пробега и связал её с вязкостью и теплопроводностью газа. Примерно в то же время Л. Больцман обобщил распределение Максвелла на случай, когда газ находится во внешнем поле, доказал теорему о распределении энергии по степеням свободы, вывел кинетическое уравнение, дал статистическое истолкование энтропии и показал, что закон её возрастания является следствием кинетического уравнения. Построение классической С. ф. было завершено к 1902 в работах Дж. Гиббса. Теория флуктуаций была развита в 1905—06 в работах М. Смолуховского и А. Эйнштейна. В 1900 М. Планк вывел закон распределения энергии в спектре излучения чёрного тела, положив начало развитию как квантовой механики, так и квантовой С. ф. В 1924 Ш. Бозе нашёл распределение по импульсам световых квантов и связал его с распределением Планка. А. Эйнштейн обобщил распределение Бозе на газы с заданным числом частиц. Э. Ферми в 1925 получил функцию распределения частиц, подчиняющихся принципу Паули, а П. А. М. Дирак установил связь этого распределения и распределения Бозе — Эйнштейна с математическим аппаратом квантовой механики. Дальнейшее развитие С. ф. в 20 в. шло под знаком приложения её основных принципов к исследованию конкретных проблем.
Лит.: классические труды: Больцман Л., Лекции по теории газов, пер. с нем., М., 1956; его же, Статьи и речи, [пер. с нем.], М., 1970; Гиббс Дж. В., Основные принципы статистической механики, пер. с англ., М. — Л., 1946. Учебники: Ансельм А. И., Основы статистической физики и термодинамики, М., 1973; Леонтович М. А., Статистическая физика, М. — Л., 1944: Ландау Л. Д., Лифшиц Е. М., Теоретическая физика, т. 5, 2 изд., М., 1964; Майер Дж., Гепперт-Майер М., Статистическая механика, пер. с англ., М., 1952; Киттель Ч., Квантовая теория твердых тел, пер. с англ., М., 1967; Хилл Т., Статистическая механика. Принципы и избранные приложения, пер. с англ., М., 1960; Хуанг К., Статистическая механика, пер. с англ., М., 1966. Литература по специальным вопросам: Абрикосов А. А., Горьков Л. П., Дзялошинский И. Е., Методы квантовой теории поля в статистической физике, М., 1962; Боголюбов Н. Н., Проблемы динамической теории в статистической физике. М. — Л., 1946: Гуревич Л. Э., Основы физической кинетики, Л. — М., 1940; Силин В. П., Введение в кинетическую теорию газов, М., 1971; Физика простых жидкостей. Сб., пер. с англ., М., 1971.
Л. П. Питаевский.

Рис. 1. Зависимость вращательной свр (а) и колебательной (б) частей теплоемкости двухатомного газа (в единицах классических значений теплоемкости) от температуры Т.

Рис. 2. Функция распределения Ферми — Дирака.

Рис. 3. Сравнение функций распределения Максквелла (М), Ферми — Дирака (Ф. — Д.) и Бозе — Эйнштейна (Б. — Э.). По оси координат отложено число частиц на одно состояние с энергией e.